
IMDRF Codes: The Key to Standardized Medical Device Data Management
As an internationally standardized system, the IMDRF codes ensure clear communication between medical device manufacturers and regulatory authorities. They provide the basis for transparency and cooperation on a global scale.
But how are the codes structured? What is the content behind them? And most importantly, how and where can these codes be applied in practice? In this blog post, we will take a closer look at the IMDRF codes, explain how they are used, and give you valuable tips on how to use them.
As a special highlight, we would like to introduce you to our user-friendly web application, which greatly simplifies the use of the IMDRF tables and ensures greater clarity. Codes can be collected and transferred to your system with a single mouse click. This not only makes work easier, but also much more efficient. We will show you how to get the most out of this application. But first, this article will cover the basics of IMDRF-Codes.
What is the IMDRF?
The IMDRF stands for International Medical Device Regulators Forum and is the successor to the Global Harmonization Task Force (GHTF).
The IMDRF is a voluntary group of medical device regulators from around the world who have come together to achieve international convergence and harmonization of medical device regulations. The IMDRF is organized into working groups that address and publish documents on various topics.
Current members are Australia, Brazil, Canada, China, the European Union, Japan, Russia, Singapore, South Korea, the United Kingdom and the United States.
For more information, visit the official IMDRF website.
IMDRF Codes – Adverse Event Reporting (AER)
The aim of the IMDRF Adverse Event Terminology Working Group is to develop and support a harmonized terminology and a standardized system for describing information about medical device incidents and correlations.
This system consists of alphanumeric codes developed primarily for medical device manufacturers (reporters) and regulatory authorities to facilitate the reporting of adverse events.
Why can IMDRF codes help?
When an adverse event occurs with a medical device on the market, it must be reported according to various regulations. The event descriptions usually contain a lot of text describing what happened, how a device was involved in the event, how the event affected the patient, user or third party, what actions may have been taken, etc. Depending on the author, different terms, formulations and explanations of the facts mean that reported incidents are not directly comparable and therefore cannot be used for analysis.
In order to summarize and standardize these usually extensive descriptions, individual text modules are converted into IMDRF codes. This has the following benefits
- Concise and accurate recording and reporting of medical device incidents
- Reduce ambiguity and increase the efficiency of the assessment process
- Use to identify potential new risks and for trend analysis, enabling faster response by regulators and manufacturers
- Enable regulators to use databases or other electronic systems to monitor and analyze adverse events, thereby improving patient and public health protection.
IMDRF Codes Application Areas: An Overview
The codes of the International Medical Device Regulators Forum (IMDRF) are an essential tool for standardized communication in the field of medical devices. They are used not only for efficient communication between manufacturers and regulators, but also in various areas of the medical device lifecycle. The main application areas are:
Communication with Regulators
An important application of the IMDRF Codes is reporting to regulatory authorities. Serious incidents and adverse events are reported in the Manufacturer Incident Report (MIR) (DocsRoom – European Commission). Within this form, you are asked to provide specific information about problems with medical devices (section “3.2 Information about problems with medical devices”). They also help to identify and group similar incidents (Section “4.3.1 Use of IMDRF terms and codes to identifying similar incidents”). This promotes a clear and consistent presentation of incidents and simplifies analysis.
Post Market Surveillance (PMS)
Once a medical device is on the market, product safety monitoring is essential. The use of IMDRF codes also plays a key role in Post Market Surveillance (PMS). According to the MDCG Guidance 2022-21 (Guidance – MDCG endorsed documents and other guidance – European Commission), these codes should be used in the Periodic Safety Update Reports (PSUR) for a consistent and comprehensible presentation of incident reports. They allow a structured evaluation of safety-related data and support the continuous improvement process of medical devices.
Internal use: trend analysis and quality management
Manufacturer codes can also be used effectively internally. They provide a standardized basis for trend analysis to identify recurring patterns of problems or incidents. The codes can be used to systematically categorize anomalies and derive actions to improve product quality and safety.
Structure of the IMDRF Codes
General structure
The structure of adverse events consists of four main groups:
- Medical Device Problem: Observations at the medical device level
- Cause investigation: Investigations into possible causes of the event and causal relationships between use of the device (whether malfunctioning or not) and adverse health effects.
- Health effects: Observations at the subject level, i.e., typically adverse health effects in patients, users or third parties.
- Component: Observations on its components, including accessories
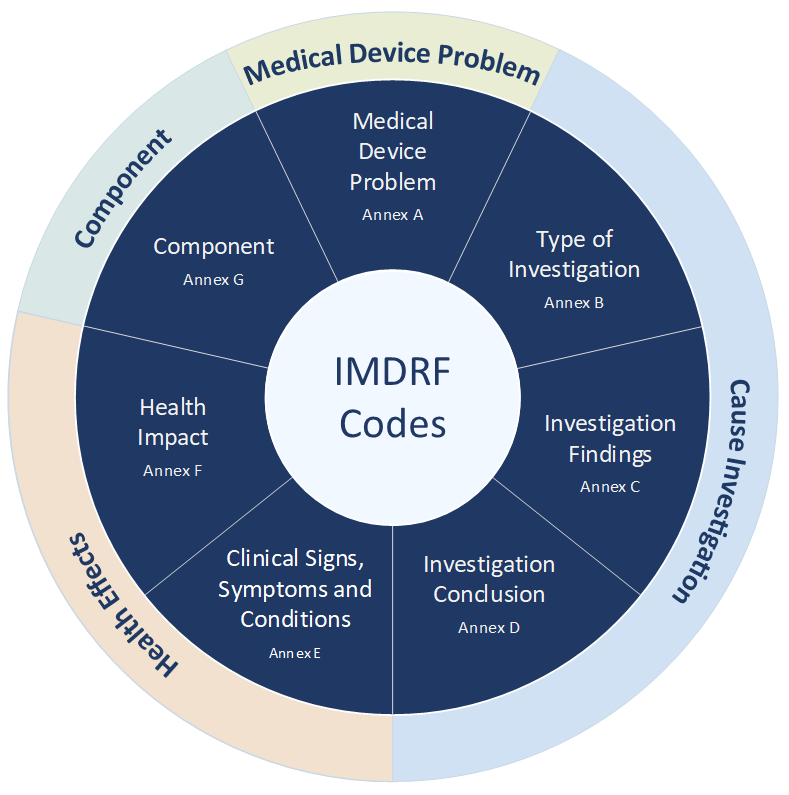
In addition, there are seven annexes developed by the working group and divided into four main groups (see Figure 1):
- Annex A: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Medical Device Problem
- Annex B: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Type of Investigation
- Annex C: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Investigation Findings
- Annex D: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Investigation Conclusion
- Annex E: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Health Effects – Clinical Signs and Symptoms or Conditions
- Annex F: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Health Effects – Health Impact
- Annex G: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Medical Device Component
A detailed explanation and breakdown of the annexes with additional examples can be found below in the article (Link). Alternatively, information can be found in the document “IMDRF terminologies for categorized Adverse Event Reporting (AER): terms, terminology structure and codes” in Section “4.5 Description of the four sets of terminologies” Terminologies for Categorized Adverse Event Reporting (AER): terms, terminology and codes | International Medical Device Regulators Forum prepared by the Working Group.
Structure and explanation of the individual levels
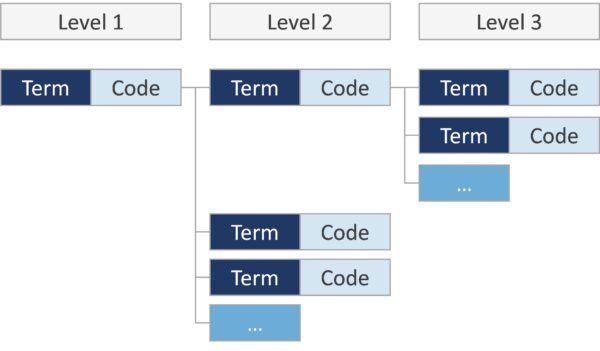
In each annex the codes are presented in a hierarchical codes structure.
General, overarching terms form the entry level (level 1), with levels 2 and 3 providing more detail and describing the general term at level 1 in more detail.
The four key terms mentioned (term/terminology, code and hierarchical code structure) are briefly explained below:
- Term/Terminology: The use of terminologies (i.e. a controlled set of well-defined terms) can aid in the description of events by reducing ambiguity of narrative text through categorization of events.
- Code/coding: Ambiguity can be further reduced by the use of alphanumerical codes, assigned to a predefined term from a given pre-defined and controlled terminology. The assignment of these codes is known as “coding”.
- Hierarchical coding structure refers to the logical arrangement of such coded terms in branching structures comprising several levels, i.e. comparable to a logical decision tree.
A line consists of two main components: the term/terminology, which is written as text, and the alphanumeric code. Each term can be uniquely identified by the code.
Structure and explanation of the codes
The Codes are structured as follows:

A code always begins with a capital letter (A-G), which indicates the system to which the code belongs. This is followed by the alphanumeric code, which consists of two digits (nn[nn][nn]). The sequence of digits can therefore consist of two, four or six digits. The number of pairs of digits indicates the level of the code. The more digits a code contains, the more detailed the information.
Explanation of the table structure
Where can I find the terms and codes for the annexes? The working group has created a table for each Annex A-G. These tables can be downloaded as JSON or XLSX from the IMDRF website: Terminologies for Categorized Adverse Event Reporting (AER): terms, terminology and codes | International Medical Device Regulators Forum. The structure of these tables is explained below.
Information Field
At the beginning of each table there is an info box with the following information:

1. Annex Name
2. Annex Title
3. Release Number
4. Annex Description
5. Annex Instruction
6. Annex Approval Date
Terms, Terminologies and Codes
Below the info box, the terms are divided into levels (1-3) as described above. Each term at each level is assigned a unique alphanumeric code. There are exceptions to the number of levels in Annexes B and D. In Appendix B there is only Level 1 and in Annex D there is Level 1 and Level 2.
In this example from Annex A, the medical device problem is an electrical/electronic problem (Level 1). Level 2 indicates what the electrical problem might be, in this example a battery problem. The third level (Level 3) describes the battery problem in more detail, for example, Failure of Battery Operation (A070503).
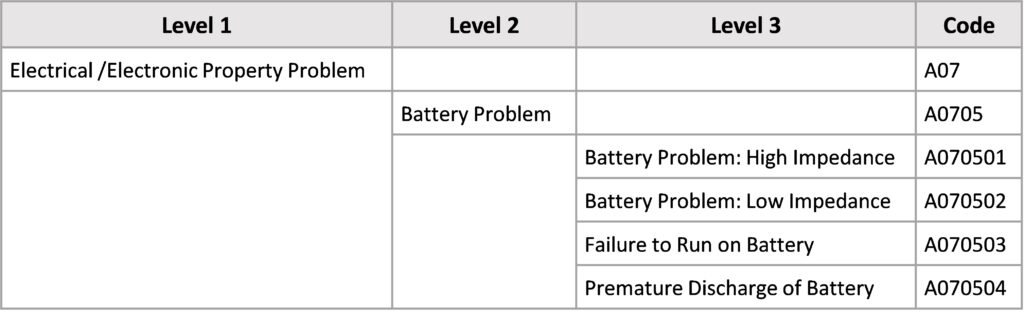
The hierarchical structure shows exactly which case it is. From the codes, the following information can be determined
- Which annex the code refers to (Annex A in the example)
- The level to which the code refers – how detailed was the incident described
Descriptions, Definitions and Examples
In addition, definitions are provided for the terms of the various levels and codes to explain the terms. Below is an example of the descriptions of codes A01, A0101 and A010101

The other columns provide additional information on the status and history of each term.
A change log and release notes for the latest version of the tables are also available on the IMDRF website: Terminologies for Categorized Adverse Event Reporting (AER): terms, terminology and codes | International Medical Device Regulators Forum.
Table Updates
Due to the nature of the medical device industry and the introduction of new technologies, materials, designs, procedures, etc., it is expected that the terminology for medical devices and related components will need to be updated to reflect technical advances. Therefore, regular review and maintenance of terminology and codes is required to add, modify or delete terms as necessary. When terms become obsolete, these obsolete terms and their corresponding codes are not deleted, but are marked as “inactive”. These obsolete terms are available for reference only and cannot be selected. There are two ways to handle inactive codes:
1) Re-coding – Adapt all codes in the internal system
2) Set a transition date – time when a new coding will take effect.
The first option is not recommended. Re-coding requires significant effort and may result in additional regulatory reporting. If a transition date is introduced, reports may need to be adjusted to reflect the new codes, but otherwise no additional work is required.
Change requests can be submitted by September 1 of each year. The update is published in March of the following year.
All changes are listed in Annex B: IMDRF terminologies for categorized Adverse Event Reporting (AER) – Change Log. Change requests cannot be submitted by individuals, but must go through national competent authorities or stakeholders. Further information can be found on the following IMDRF webpage IMDRF Adverse Event Terminology Maintenance | International Medical Device Regulators Forum.
Further Explanation of Annexes A-F
Medical Device Problem terms/codes (Annex A – A XX|XX|XX):
These terms provide a way to record the problems encountered at the device level through observations, but without describing the possible reasons or causes for the observed problems or failures. Annex A contains a comprehensive, hierarchically structured list of terms and codes for medical device problems. The terms and codes at this level are shown as examples in Figure 5.
Cause investigation terms/codes (Annex B-D):
Type of Investigation terms/codes (Annex B – B XX): Annex B specifies what was investigated to determine the type of incident. In this annex, there are only Level 1 terms. For example, if the production records were examined, the term “Analysis of Production Records” – B14 can be selected.
Investigation Findings terms/codes (Annex C – C XX|XX|XX): Annex C contains the results of the specific investigation that is key to determining the cause. Like Annex A, this annex is organized hierarchically. Looking at the example above for Annex B (B14), the result of the investigation could be that there were problems during the manufacturing process, such as assembly problems (Assembly Problem Identified – C1601).
Investigation Conclusion (Annex D – D XX|XX): Annex D contains the conclusions of the investigation. The conclusions state the cause of each adverse event. This annex is also hierarchical, but only two levels. A possible conclusion here could be that the event was due to a problem in the manufacturing process (Manufacturing Deficiency – D0103) and it could also have been caused by inadequate training (Cause Traced to Training – D08).
Health Effects terms/codes (Annex E and F): These terms are used to describe the observed signs and symptoms and outcomes associated with a medical device adverse event. Diagnostic characteristics are not used. Annex E contains a list of clinical signs, symptoms and conditions that is detailed enough to capture the health implications for reporting adverse events or incidents related to medical devices. At the same time, it is general enough to avoid the creation of further comprehensive terminology systems. Annex F contains a list of possible consequences and effects of the event. The terms from these two annexes together provide a description that covers both the clinical observation and the impact on the affected person.
Clinical Signs, Symptoms and Conditions terms/codes (Annex E – E XX|XX|XX): Annex E contains terminology to describe the observed condition of affected individuals in the context of a medical device incident. These terms should not be used to describe signs, symptoms and conditions that existed before the incident. Due to the close collaboration between IMDRF and MedDRA, these terms are closely aligned with a subset of MedDRA terms. A mapping of the IMDRF codes to the MedDRA terms can be found in the table in Annex E. This annex is categorized by organ system and physiological problem. Some terms appear in more than one category to make it easier to find the correct term. In these cases, each repeating term is assigned only a unique code based on its primary category.
Health Impact terms/codes (Annex F – F XX|XX|XX): Annex F provides terminology to describe the consequences to the affected person resulting from a medical device adverse event. The resulting consequences may include the final outcome for the patient and/or the interventions or procedures required based on the clinical signs, symptoms and conditions listed in Annex E.
Multiple codes may also be used if different actions or interventions were required, for example: an investigation that had to be performed in response to the event.
If a code from Annex E has been assigned, a code from Annex F does not necessarily have to be assigned.
Component terms/codes (Annex G – G XX|XXX|XX): These terms can be used to identify the specific part or component of the medical device involved in the incident. The component terms are grouped into sections to make it easier for the user to find an appropriate term. Each term appears in only one section, and some simple rules have been applied to select the appropriate section in cases where a term could logically fit in more than one section. Components with multiple attributes have been assigned to a section in the following order Safety > Measurement > Optical > Biological & Chemical > Electrical & Magnetic > Mechanical. There will be some devices and/or incidents for which it is not necessary or appropriate to select a single component.
How to Use the IMDRF Tables
Using the IMDRF tables to document incidents may seem complex at first glance. With seven tables and a multitude of terms, it is easy to lose track. However, with a few focused approaches, the process can be greatly simplified. Below are some helpful tips for using the tables efficiently.
Getting an overview and sorting
The first step in successfully using the tables is to become familiar with their structure and contents. It is important to go through the entire contents of the tables to get an overview. It is especially helpful to sort the terms in the tables based on the company’s own product. In this way, it is possible to eliminate obviously irrelevant codes at an early stage. An example: For a classic infusion set without electronic components, terms or codes associated with electric shocks can be excluded.
Using the search function: synonyms and alternative terms
When an incident is reported, the best case scenario is that the report already contains some important information, such as
- What happened?
- Who and what products were involved in the incident?
- Was anyone injured?
- How did the incident affect the person(s) involved?
Based on this information, it is possible, and in some cases beneficial, to use the search function. When searching for appropriate codes, it is helpful not to stick strictly to the terms listed. Instead, synonyms or alternative phrases may be considered.
Detailed description with precise codes
When documenting an incident, the more detailed the description, the better. Whenever possible, Level 3 codes should be used, as they provide the highest level of detail. If sufficient information is not available, Level 2 or Level 1 codes can be used. In some cases, it may be useful to use multiple codes within the same annex to describe the event more comprehensively.
No appropriate terms or insufficient information
If neither an appropriate term nor sufficient information is available and the level 1 terms cannot be used, the tables still offer possible solutions. There are specific codes for such cases, e.g.:

These codes provide a way to adequately document incidents that are incomplete or cannot be clearly assigned.
Standardized code sets for recurring events
For recurring events, documentation can be made much more efficient by using fixed code sets. Defining such standardized codes ensures uniform and consistent documentation, which not only saves time, but also makes it easier to evaluate the data internally.
Special features of each annex
Annex E: This annex contains very specific level 3 terms. If there is any uncertainty about the assignment, it is advisable to consult the relevant expertise to select the correct terms. A precise code adds value and increases the informative value of the documentation.
Annex G: Especially in Annex G, it is advisable to use the search function to look for relevant terms or components. This can speed up the process considerably, especially for more complex products.
Case Study
The following case study is intended to illustrate the use of the IMDRF codes and is based on a fictitious scenario. The message shown is deliberately described in great detail in order to provide as much information as possible from which to select the appropriate codes – in reality, such messages are usually much shorter and relevant information must usually be requested first.
The codes listed are examples of options that may be used depending on the root cause investigation performed and the findings obtained. Depending on the specific circumstances of the case, other IMDRF codes may be applicable. The case study is intended to stimulate reflection on the many possible applications of the tables and to make an appropriate selection for individual scenarios.

Based on the electronic blood pressure monitor incident described, and using IMDRF adverse event reporting terminology, the appropriate codes for Appendices A-G could be as follows:
Annex A: Medical Device Problem Terms and Codes
- Code: A1406
- Term: Inflation problem
- Description: Problem associated with the inability of the device to expand or enlarge with the intended inflation agent (e.g. saline or air).
- Rationale: Problems when inflating the cuff, e.g. incomplete or excessive inflation.
Annex B: Cause Investigation – Type of Investigation Terms and Codes
- Code: B01
- Term: Testing of Actual/Suspected Device
- Description: The investigation employed relevant empirical testing of the actual device suspected in the reported adverse event in order to establish their functional and other properties and to identify possible causes for the adverse event. Relevant testing would typically be based on test methods used for evaluating safety and performance as described in the latest relevant standards.
- Rationale: The actual device affected by the event would need to be tested to determine functionality and possible causes of the problem (e.g., mechanical or electronic).
- Code: B14
- Term: Analysis of Production Records
- Description: The investigation involved the analysis of relevant production records in view of supporting the identification of possible causes for the adverse event.
- Rationale: Production records should be reviewed to determine if there were any manufacturing or quality control problems that could explain the event.
- Code: B24
- Term: Event History Log Review
- Description: The investigation involved the analysis of the history log files retrieved from the device to support possible causes for the adverse event, includes device interrogation.
- Rationale: A review of the device’s history logs (if available) can provide important clues about malfunctions or user interactions prior to the incident.
Annex C: Cause Investigation – Investigation Findings Terms and Codes
- Code: C0201
- Term: Electrical/Electronic Component Problem Identified
- Description: The performance of an electrical or electronic component was found to be inadequate.
- Rationale: Failure of a component of the device that caused the described problem.
Annex D: Cause Investigation – Investigation Conclusion Terms and Codes
- Code: D02
- Term: Cause Traced to Component Failure
- Description: Expected or random component failure without any design or manufacturing issue.
- Rationale: The problem with the cuff may have been caused by the failure of an electrical or mechanical component. Since the air did not escape even though the device was turned off, this could indicate a malfunction in the valve system or control electronics.
- Code: D0102
- Term: Human Factors Engineering – Device Difficult to Operate
- Description: Problems traced to inappropriate and/or inadequate design of the user interface or usability of the device.
- Rationale: If the device did not provide a way to manually release the excessive pressure, or if the user guidance was inadequate, this could be due to insufficient consideration of human factors in the design.
- Code: D0105
- Term: Missing or Inadequate Safety Measures
- Description: Problems traced to inadequate design or complete lack of safety features and/or protective measures.
- Rationale: If the device has no safety mechanisms to automatically relieve excessive pressure or allow emergency release, this code would be appropriate.
- Code: D0108
- Term: Inadequate Software Design
- Description: Cause traced to inadequate or inappropriate design of the device software resulting in device malfunction
- Rationale: As the device displayed an error message (“E04 – Cuff Pressure Too High”), but still did not deflate, there may be a software malfunction that is blocking the deflation.
Annex E: Health Effects – Clinical Signs, Symptoms and Conditions Terms and Codes
- Code: E2330
- Term: Pain
- Description: An unpleasant sensory and emotional experience that is associated with actual or potential tissue damage or described in such terms.
- Rationale: The patient felt pain in her arm due to the constant pressure of the cuff.
Annex F: Health Effects – Health Impact Terms and Codes
- Code: F26
- Term: No health consequences or impact
- Description: No apparent harm occurred in relation to the adverse event.
- Rationale: There were no long-term health consequences for the patient.
Annex G: Medical Device Component Terms and Codes
- Code: G02027
- Term: Power Supply
- Definition: Component designed to supply electrical power to a device or system.
- Rationale: If the blood pressure monitor had a faulty power supply (e.g., voltage problems), this could have caused the control electronics to malfunction and prevent air from escaping.
- Code: G02026
- Term: Power Cord
- Definition: A flexible cable designed to connect an electrical device to a power source.
- Rationale: A loose contact or insufficient power may have caused the unit to behave in an uncontrolled manner, causing the valve to malfunction.
- Code: G02004
- Term: Cable, Electrical
- Definition: A long, thin, multistranded electrical wire designed to conduct electricity within or between devices.
- Rationale: If internal wiring is defective, control of the air release may be compromised.
- Code: G0203401
- Term: Power Switch
- Definition: A switch designed to regulate the power to a device or a specific function within a device.
- Rationale: If the unit was not turned off properly or the switch was defective, the air release may not work.
Once you have learned the basics of IMDRF codes, you can start using the application right away. In this article (link) you will find all the information about our web application.




